
Technology
Heterogeneity is defined as the quality or state of being diverse in character or content. Heterogeneity drives our diversity as humans via a set of variations in our DNA, resulting in the differences we see in our attributes, senses and ability to live a long, healthy life.
In addition to the genetic variation encoded at birth, changes to our basic genetic blueprint happen throughout our lives. These include changes on the level of single nucleotides (SNPs or SNVs), as well as larger, structural changes —such as chromosomes swapping, duplicating, or losing large sections of genetic material. These modifications affect the way our cells behave and their ability to replicate and survive. BioSkryb enables the study of this process of cellular evolution through a revolutionary new method for whole genome amplification (WGA), called Primary Template-directed Amplification (PTA)1.
WGA makes it possible to perform genomic analysis from single cells and other minute biological specimens. Commonly used methods, such as Degenerate Oligonucleotide-Primed PCR (DOP-PCR),2 Multiple Displacement Amplification (MDA),3 Multiple Annealing and Looping Based Amplification Cycles (MALBAC)4, and Linear Amplification via Transposon Insertion (LIANTI),5 have drawbacks such as amplification bias, poor uniformity, errors and artifacts, low genome coverage, inability to address all variant classes, low accuracy, poor reproducibility, and/or complex protocols that are difficult to automate or scale. PTA is a novel, isothermal WGA method that reproducibly captures >95% of the genomes of single cells, in a controlled and more uniform and accurate manner than existing approaches.6 This improves variant calling sensitivity and specificity, lowers sequencing costs, and facilitates bioinformatic analysis.

PTA offers superior coverage and uniformity in single cell WGS. WGA was performed with the PTA-based ResolveDNA® WGA Kit (first kit version, left) or with single-cell MDA (right). Plots show a portion of chromosome 1 (100 kb bins). The central area (poorly covered with both methods) corresponds to the centromere.
PTA is a core technology that enables a wide variety of emerging single-cell genomics applications, such as:
- Accurate variant analysis (SNPs, indels, SNVs and CNVs) of single cells and sub-nanogram DNA samples
- Quantitative, genome-wide assessment of CRISPR/Cas9 on- and off-target gene editing at single-cell resolution
- Genome assembly and characterization of rare and unculturable microbes
PTA takes advantage of the processivity, strand displacement activity, and low-error rate of phi29 DNA polymerase. An innovative reaction setup employs exonuclease-resistant terminators to create relatively short, double-stranded amplification products that are poor templates for subsequent cycles of amplification. This transforms the reaction from an exponential into a quasi-linear process, during which the majority of amplicons are generated from the primary template. Unlike MDA2, PTA limits the exponential propagation of priming and amplification biases, allelic skewing and other errors from daughter molecules. This results in improved uniformity, coverage and reproducibility, and significantly improved variant call rates.6
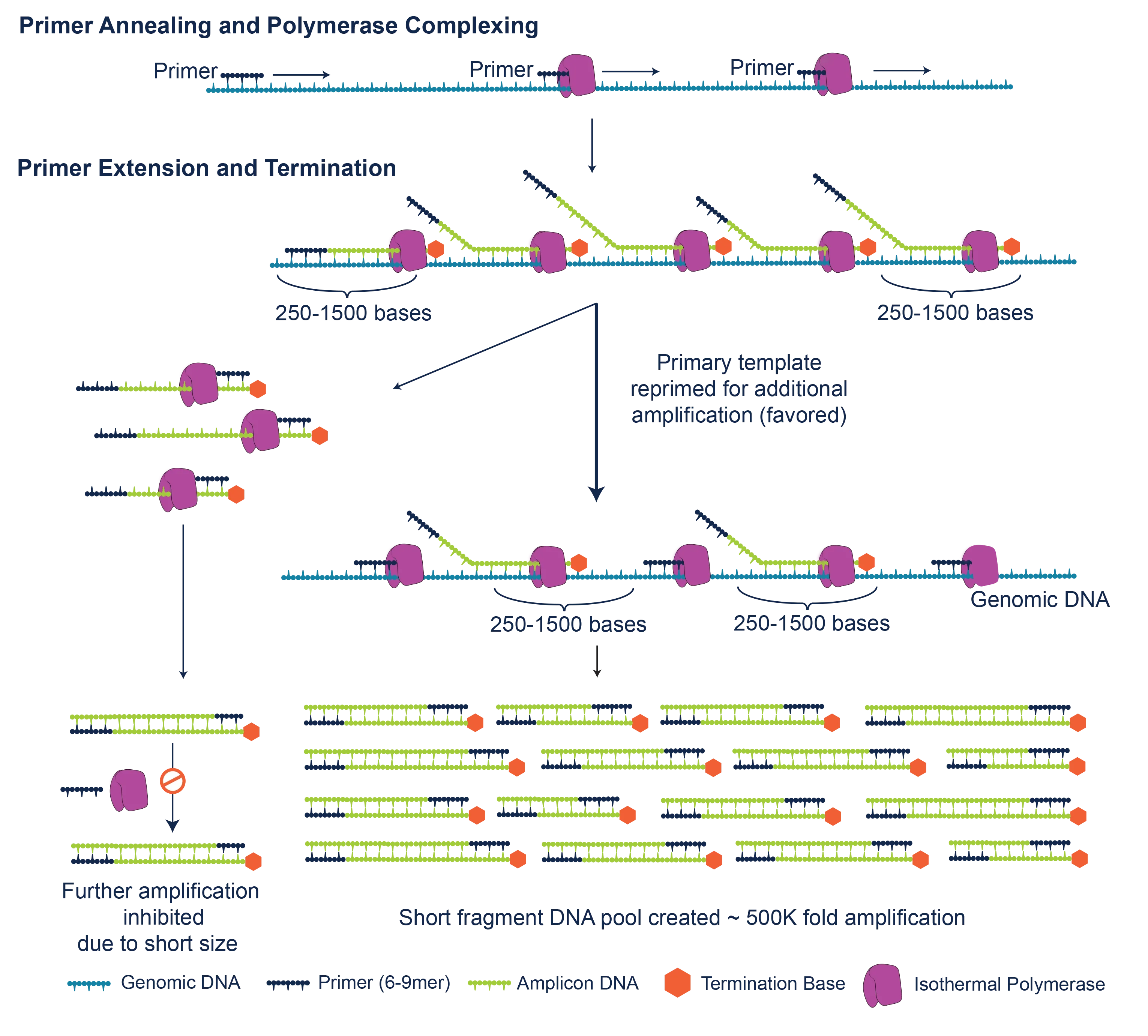
How PTA works. PTA may be performed directly from single cells (collected by FACS, microfluidic or other methods), multiple cells, or ultra-low inputs of DNA (>4 pg – 10 ng). After denaturation, random primers (6- to 9-mers) are annealed. Extension with phi29 and a proprietary nucleotide pool results in amplicons of ~250 to >1,500 bp in length. The relatively small size of these amplicons favors subsequent priming off the primary template, thereby limiting the exponential propagation of biases and errors in daughter molecules. In addition, the PTA chemistry suppresses the formation of experimental artifacts such as chimeric molecules and non-specific priming.2 PTA reaction products are double-stranded and may be converted to libraries for multiplexed sequencing on Illumina® or other platforms.
References:
- Method for Nucleic Acid Amplification. WO/2019/148119.
- Telenius H, et al. Genomics 1992;13(3): 718.
- Dean FB, et al. PNAS 2002; 99(8): 5261.
- Zong C, et al. Science 2012; 338: 1622.
- Chen C, et al. Science 2017; 356: 189.
- Gonzalez-Pena V, et al. PNAS 2021; 118(24): e2024176118.