

Commentary by:
Contributors
Single-cell oncogenic mechanistic heterogeneity defined by PTA in primary Ductal Carcinoma In Situ
Primary Template-directed Amplification (PTA) is a novel single-cell whole-genome amplification (WGA) method which yields unprecedented genomic coverage and uniformity for accurate calling of single nucleotide variation (SNV) and copy number variation (CNV). PTA is employed here to genomically profile Ductal Carcinoma In Situ (DCIS) at the single cell level, revealing distinct DNA lesions and suggesting remarkably diverse mechanisms of oncogenesis between single cells.
Understanding DCIS to invasive cancer transition requires single cell genomics
DCIS is a neoplastic proliferation of ductal epithelial cells that can be a precursor to invasive breast cancer. A fundamental research mission, in addition to understanding the influence of the stromal microenvironment, is to understand cell autonomous genomic events that drive this transition to

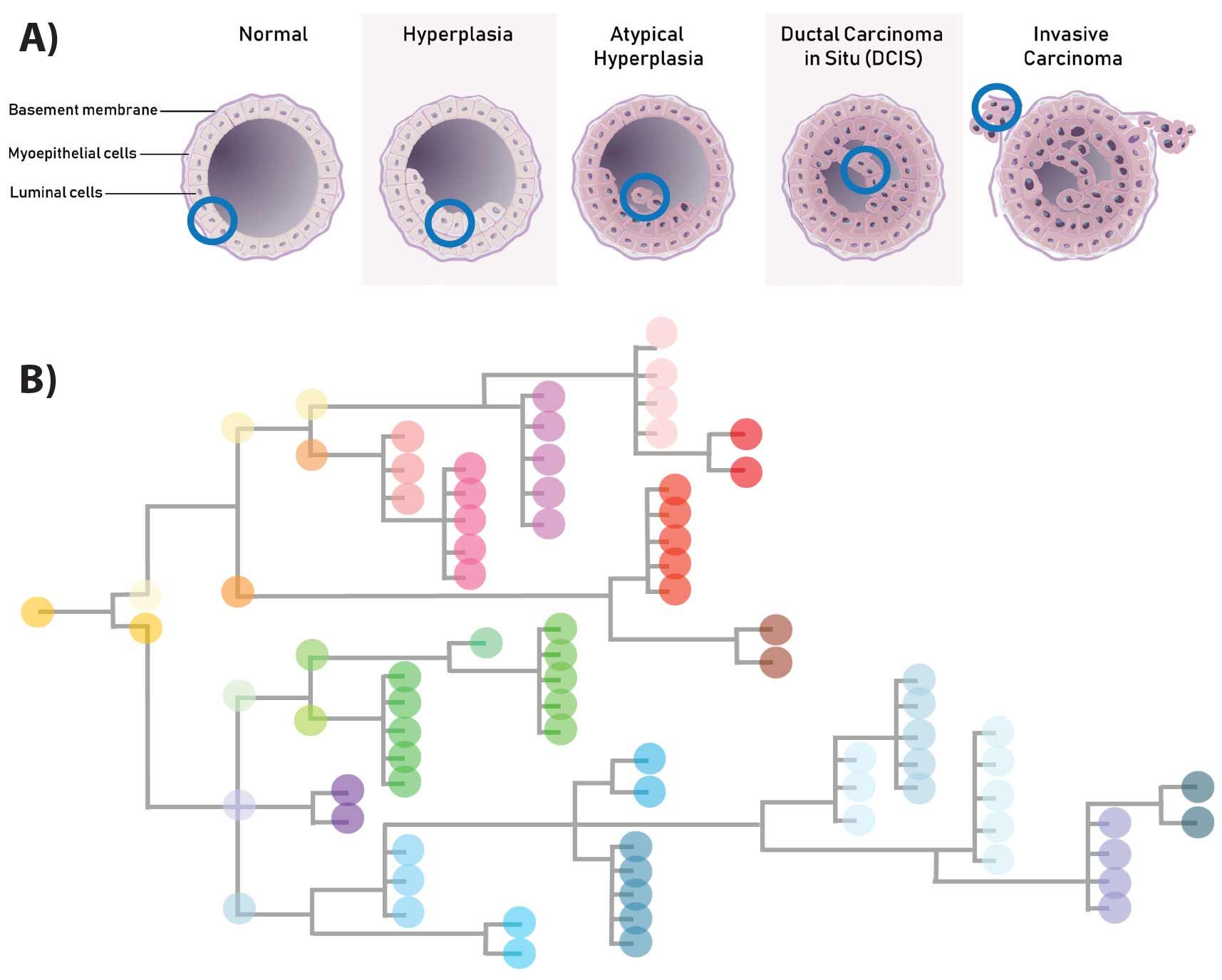
Figure 1. Ductal Carcinoma In Situ transition to an invasive state. Normal ductal epithelial cells can exist in a state of neoplasia, but be regionally confined and asymptomatic (A). Genomic lesions compound heterogeneously between single cells (B) evolving to multiple clones, some of which have the capacity to drive DCIS to become invasive and symptomatic (A).
invasive disease and to eventual metastatic dissemination of tumor cells. An "evolutionary bottleneck" is proposed (1) to select for individual tumor cells that have the genomic (and epigenomic) lesions and/or combinations of pre-existing variation leading to "bottleneck" escape from an earlier quiescent state.
These genomic profiles facilitating invasiveness are often extremely rare clones that have undergone a Volgogramesque sequence of genomic changes in a lineage progression (Figure 1). These rare clones are not detectable by conventional bulk sequencing, and thus single cell sequencing is required to illuminate these potentially actionable events. Moreover, single cell sequencing of DCIS transitioning to Invasive Ductal Carcinoma (IDC) is the only way to define the state of heterogeneity of genomic lesions that exists within the tumor.
Primary Template-directed Amplification
To faithfully capture the complete complement of genomic changes in single cells contributing to the DCIS to IDC transition, and those contributing to oncogenesis, a robust genomic amplification platform is required (2). The following categories are paramount to maximize when generating single-cell and low input amplification product for calling single nucleotide and copy number variation:
- Fraction of the genome covered
- Uniformity of genome coverage
- Allelic balance

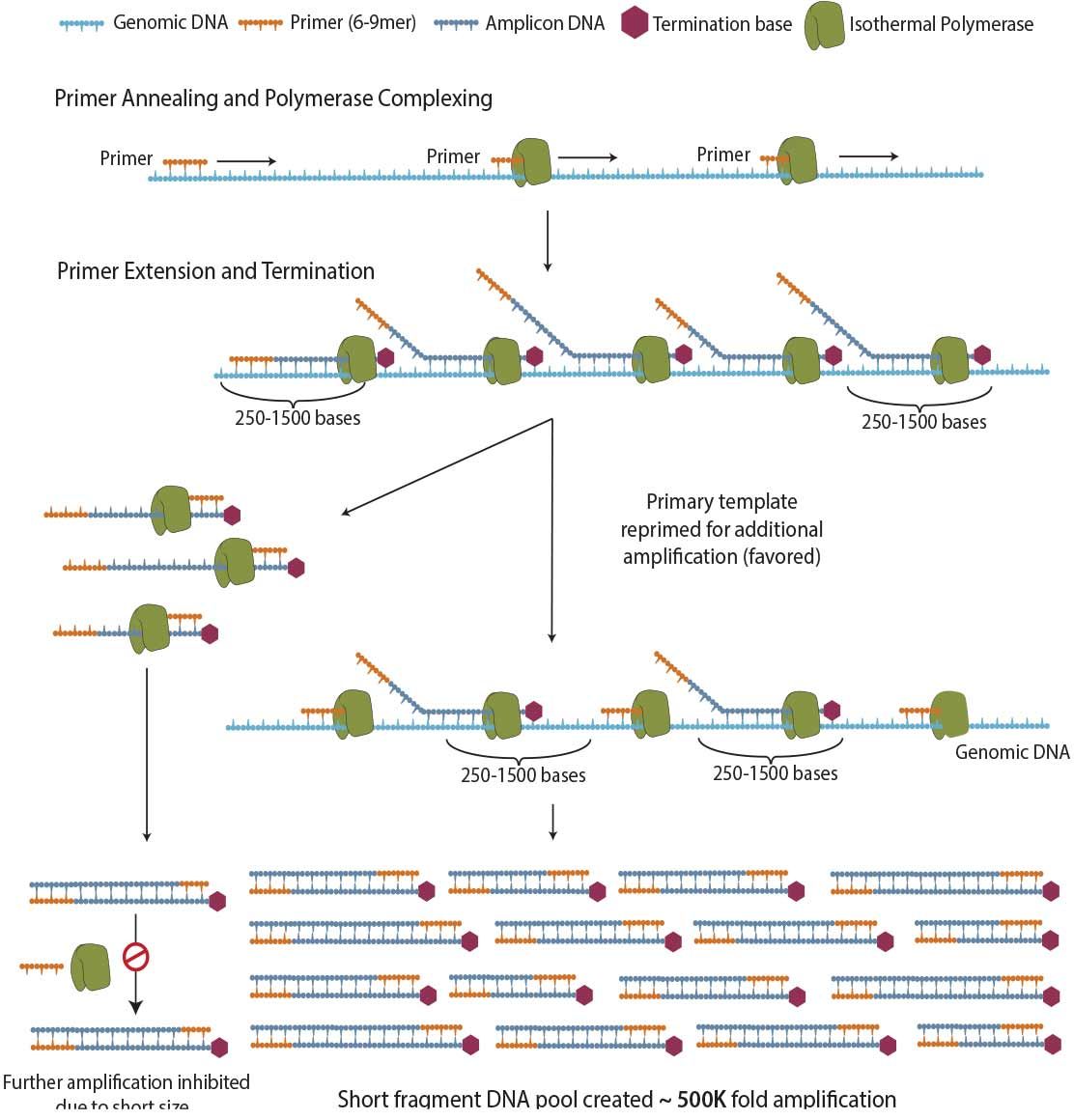
Figure 2. Schematic of PTA mechanism. ResolveDNA amplification is a random primed, isothermal polymerase driven reaction that provides unbiased amplification (2) by attenuating the size of daughter amplicons, consequentially redirecting primers to the primary template.
The concept is simple: if gaps are present in coverage, if covered regions consist of read structures dominated by peaks and valleys, or if only one allele is represented, you will fail to identify variants influencing pathology. As existing methodologies, including Multiple Displacement Amplification (MDA), have limitations in each of these categories, we have devised Primary Template-directed Amplification (PTA) (Figure 2) to surmount these issues. A proprietary amplicon termination technology limits the size of randomly-primed products, and due to the reduced propensity for these short products to re-amplify, the primers are re-directed to the DNA of interest--the primary single cell genome, not the daughter amplicons (2). This results in the phenomenon of limiting "copies of copies", yielding unprecedented ability to accurately identify genomic variation.
Patient metadata
BioSkryb Genomics collaborated with Dr. Shelley Hwang, Chief of Breast Surgery at Duke University Medical Center, to utilize a patient DCIS sample as per Duke University Medical Center's Institutional Review Board. The scope of the collaboration was to generate best-in-class single cell CNV and SNV data with ResolveDNA amplification technology and to identify genomic lesions that may be contributing to the DCIS to invasive ductal carcinoma transition. Pathologically, the patient's disease was classified as estrogen receptor / progesterone receptor positive and HER2 negative (Figure 3) and comprised both DCIS and IDC features. Clinically, the 61 year old patient was treated for DCIS in the left breast with radiotherapy and the aromatase inhibitor

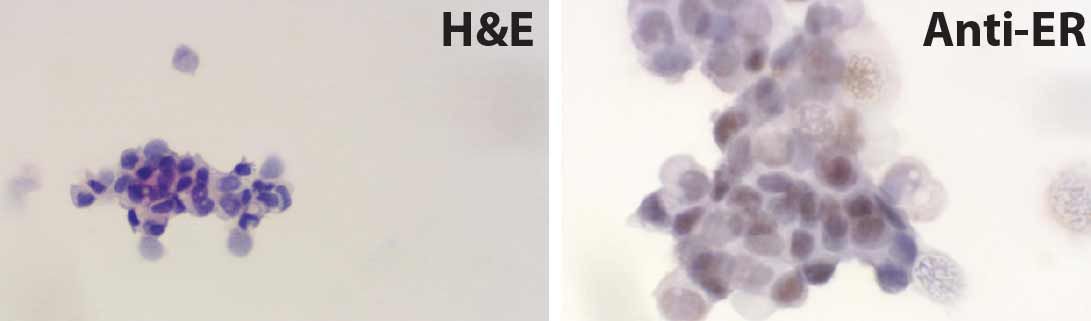
Figure 3. H&E and estrogen receptor immunostaining of primary DCIS sample. Slide smear preparation of singulated tumor cells from a patient with both DCIS and IDC histopathological presentation stained with H&E for morphological analysis. Staining was performed with the same singluated sample used for EpCAM immunoenrichment by FACS.
Arimidex. The patient had discontinued the use of Letrozole and there was no indication of recurrence.
Epithelial cell enrichment by FACS
The HER2 negative status of the tumor cells precluded the ability to enrich for ductal epithelial cells with this extracellular marker. Although the patient's tumor profiled ER/PR positive, these were not amenable as FACS markers as they are intracellular and would require chemical fixation that would interfere with PTA. Accordingly, our strategy for enrichment of ductal epithelial cells was to utilize epithelial cell adhesion molecule (EpCAM), a glycoprotein regulating adhesion and cell signaling in diverse epithelial microenvironments. We first vetted the performance of an antibody clone in cell lines; demonstrating the ability to distinguish between abundant EpCAM in the epithelial context

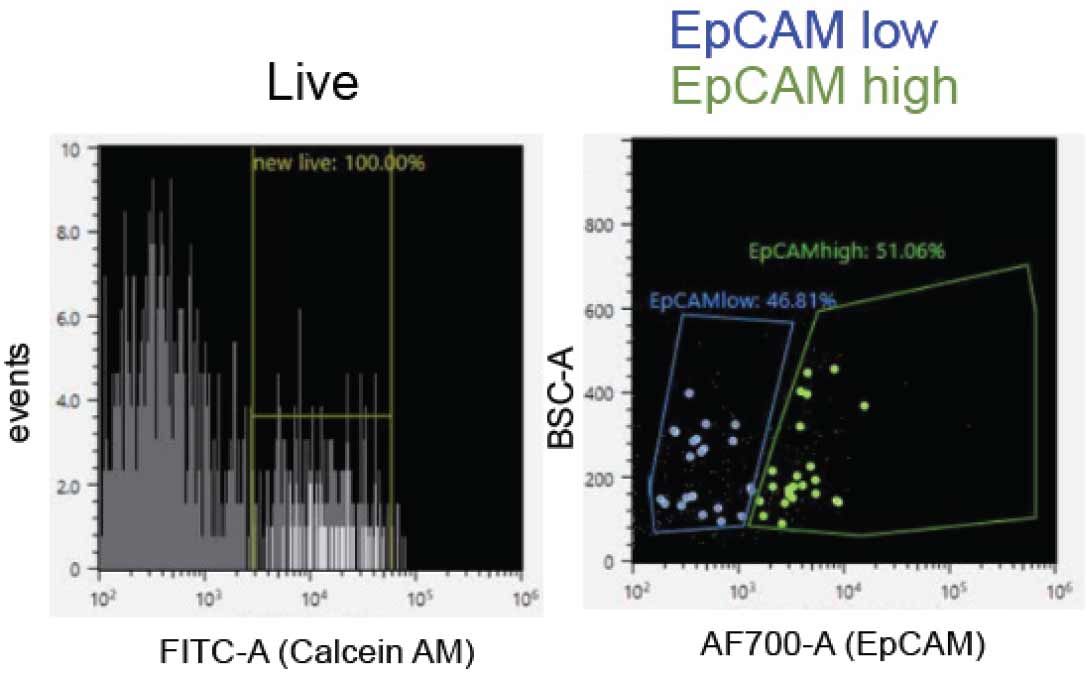
Figure 4. FACS EpCAM enrichment of primary dissociated tumor cells. Calcein-AM staining was utilized to enrich for live cells from the dissociated cell preparation derived from a surgical resection, followed by gating on EpCAM expression levels. A fluorophore-conjugated (AF700) primary antibody was employed. Single cells were then sorted into 96 well PCR plates for PTA.
of SKBR3 breast cancer cells and negligible EpCAM expression in the context of a leukemic cell line, MOLM-13. Tumor cells singulated from the primary surgical specimen were then subjected to FACS, utilizing the vetted EpCAM antibody clone and a fluorescent viability dye to enrich for live epithelial cells from a heterogeneous sample (Figure 4). Single cells were sorted directly into BioSkryb Cell Buffer in 96 well plates, to serve as a template for whole genome amplification by PTA.
ResolveDNA whole genome amplification and Illumina DNA Prep library preparation
ResolveDNA amplification (10h) of single-cell genomes was performed with 26 EpCAM-enriched DCIS/IDC singulated cells from the tumor specimen and with (5) cells from an ipsilateral biopsy of normal, adajent stromal tissue (Figure 5).

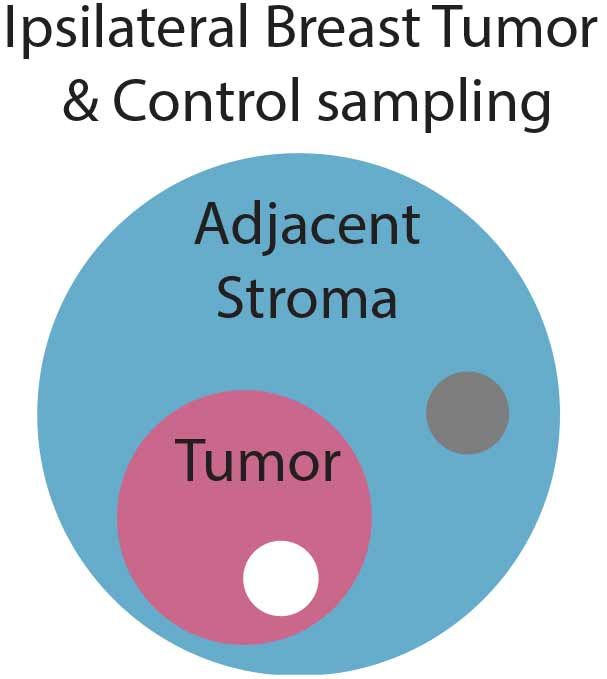
Figure 5. Tumor and stroma sampling scheme. In addition to the primary DCIS/IDC surgical resection (white circle) an adjacent stromal biopsy (gray circle) was obtained from the same breast for cell singulation; allowing tumor/stromal PTA genomic profiles to be compared.
Figure 6 highlights the uniformity of microgram-quantity amplification yield obtained from single cells and genomic DNA controls. We subsequently coupled PTA with tagmentationbased Illumina DNA prep, utilizing 100 ng of PTA product as input into the Illumina library preparation workflow.

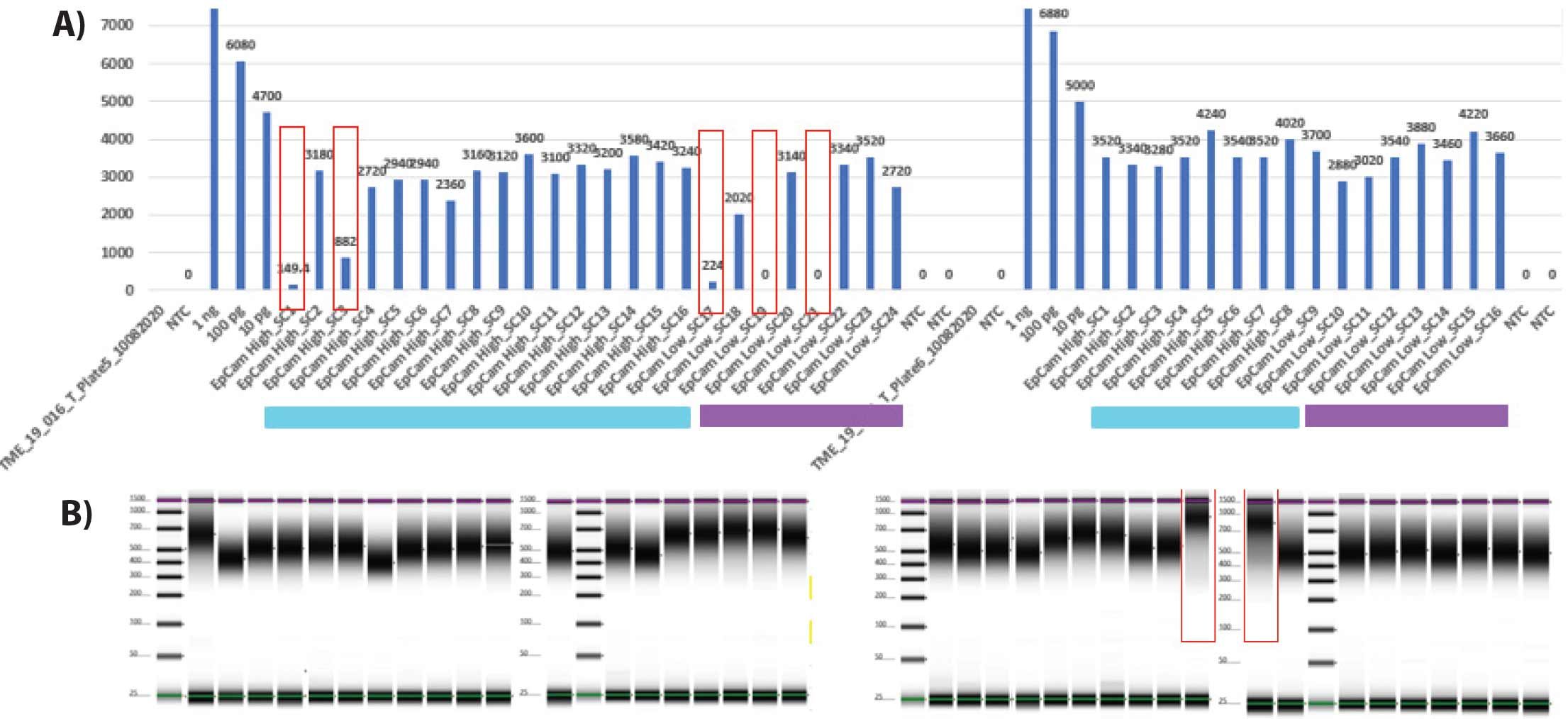
Figure 6. PTA amplification of EpCAM-enriched primary DCIS cells. In (A) EpCAM high (light blue) or EpCAM low (purple) single cells were subjected to PTA whole genome amplification reactions (top). 1 ng, 100 pg, 10 pg of GM12878 cell genomic DNA was utilized to control for reaction performance. While most cells had consistent multi-microgram PTA yield, red boxes indicate instances of no PTA yield due to FACS dropout or an instance of partial amplification potentially due to an apoptotic cell. PTA product was then utilized as direct input into the Illumina DNA Prep library preparation protocol. Tapestation sizing profiles of Illumina libraries are shown (B), with two outliers highlighted in red that were not sequenced to aberrant sizing.

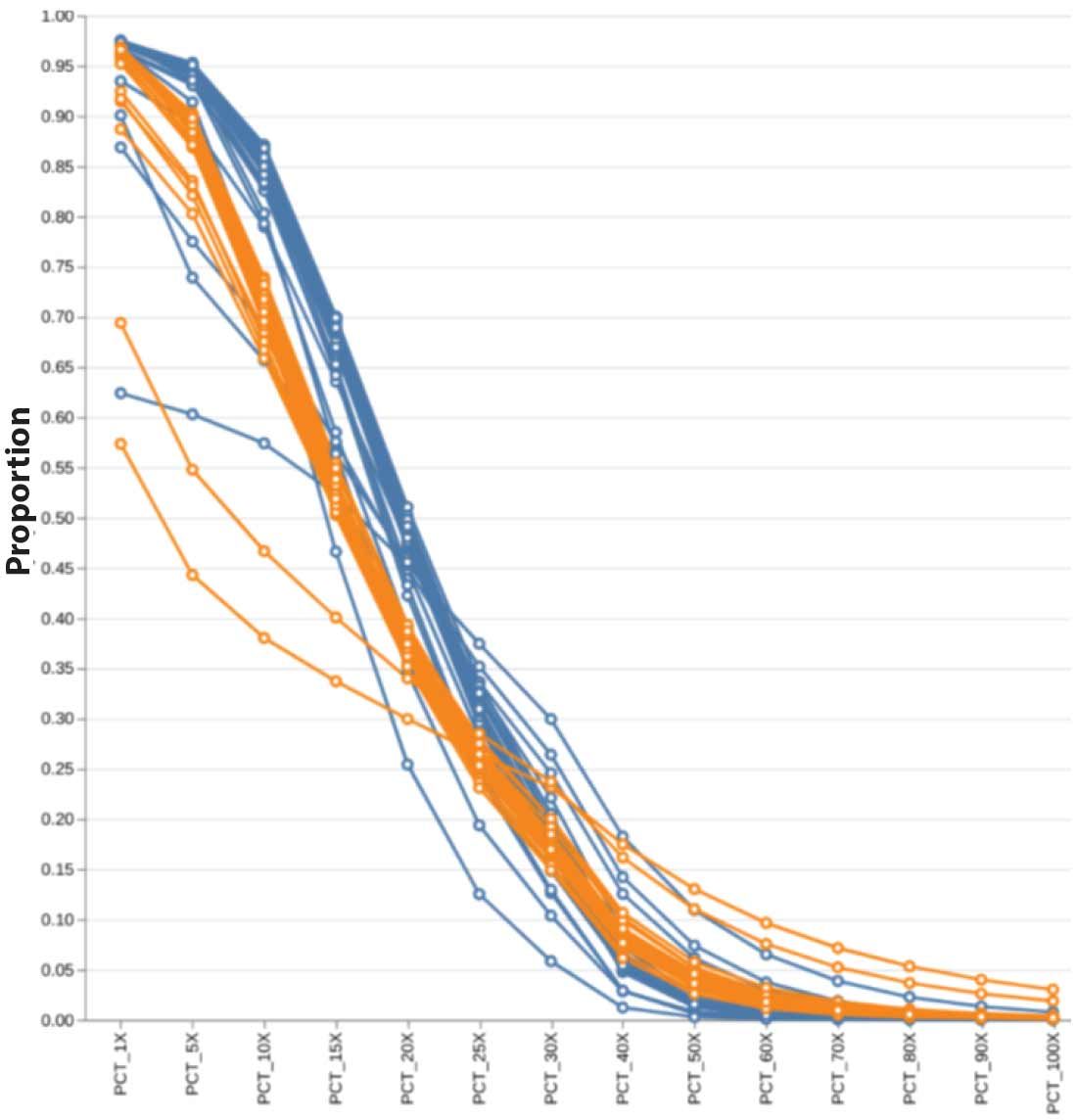
Figure 7. Genomic coverage profile of PTA-amplified DCIS/IDC and normal breast single cells. The proportion of the genome that has the indicated coverage is shown for the primary single cells (blue lines) relative to control GM12878 lymphocytes (orange). Most patient cells exceeded 95% of the genome covered by at least one read.
Sequencing and genomic coverage metrics
Illumina DNA Prep libraries representing 31 single cell PTAamplified genomes were sequenced by synthesis on a NovaSeq 6000 S4 flow cell to generate 550 M paired end, 150 bp reads. The BWA-MEM algorithm was employed for alignment to the GRCh38 genome assembly, and a panel of sequencing metrics was generated, including Preseq (4), a measure of library complexity that estimates genomic coverage and uniformity. In addition to Preseq count, we tabulated percentage of reads mapping to GRCCh38 as well as the mitochondrial read percentage, which is an indicator of the efficacy of cell lysis during the PTA workflow.
The summarized sequencing metrics (mean value + SD) are as follows for the 31 patient cells employed in this study:
Preseq count: 3.84 E9 +/- 0.495 E8
Alignment rate: 0.998 +/- 0.002
Chr. M fraction: 0.001 +/- 0.0006
Percent 1X coverage: 0.951 +/- 0.068
The preseq estimation of library complexity and low mitochondrial fraction was predictive of the robust mean genomic coverage (Figure 7 for individual cell plotting) we obtained for these patient cells.
Heterogeneity revealed: oncogenically-relevant CNV and SNV diversity in DCIS
The robust sequencing metrics and genomic coverage uniformity obtained from coupling PTA single cell genome amplification with Illumina DNA Prep provided confidence in copy number and single nucleotide variation. We employed Ginkgo and DRAGEN algorithms to call CNV and SNV, respectively. Even among a sample set of 31 individual cells, we saw remarkable intratumoral CNV diversity (Figure 8). Regional chromosome loss coincided with tumor suppressor genes known to be influential in DCIS (3), including retinoblastoma 1 (Rb1) and p53.
In addition, loss of the chromosomal region encompassing BRCA2 was observed (13q12.3), suggesting a contribution of DNA repair defects contributing to neoplasia. In addition to these prototypical DCIS chromosomal alterations (3), we importantly identified a cell harboring multiple large copy number losses (Chr. 2, 6, 8, 9, 12, 13, 16, 17) exemplifying the marked clonal heterogeneity observed within this patient tumor sample, but of which the consequences on tumor suppressor loss-of-function remain to be determined.
A fundamental power of single cell analysis is the ability to delineate cell lineage. In this specific patient tumor, the majority of single cells did not have any apparent gross CNV (Figure 8B).

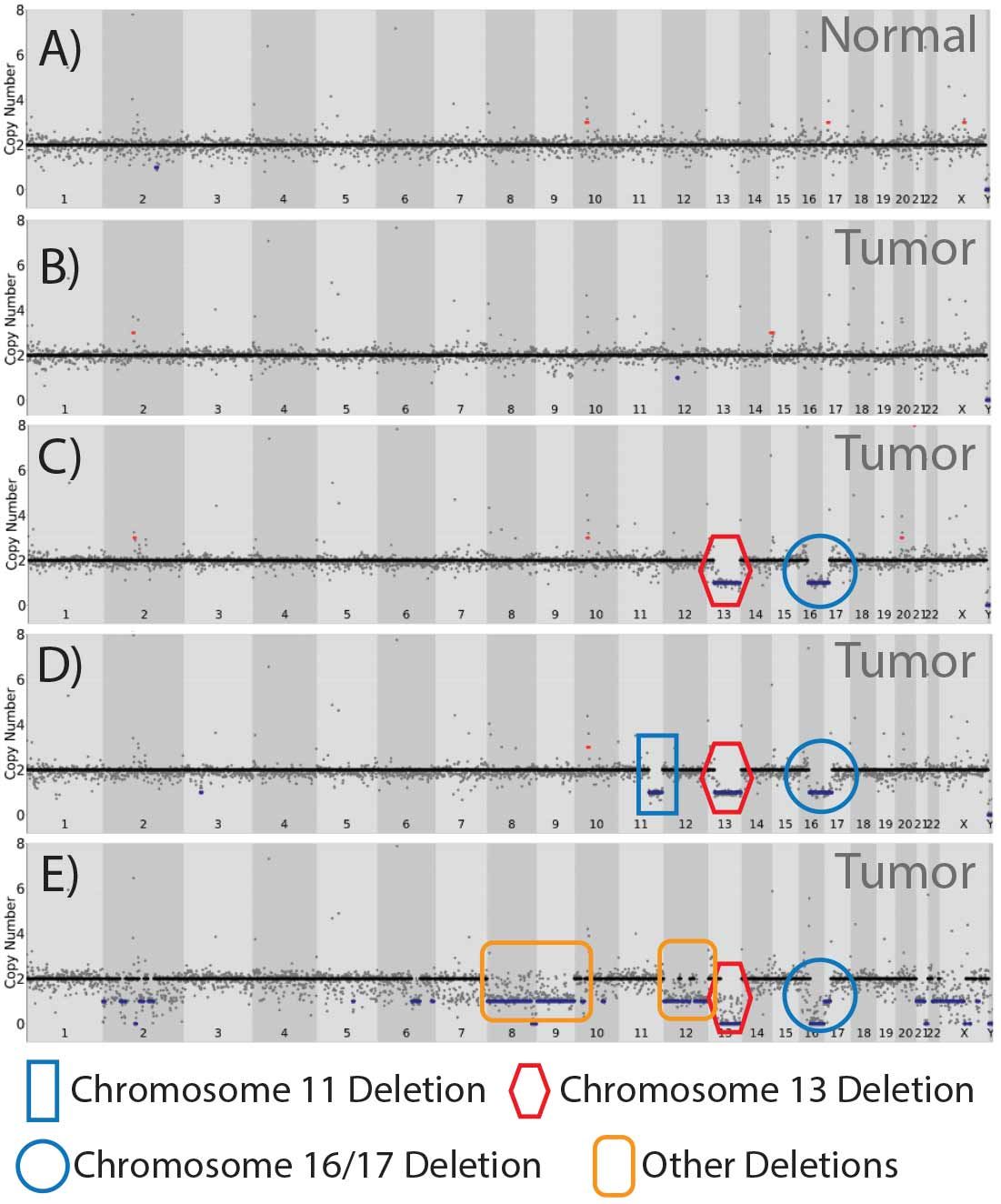
Figure 8. CNV analysis classifying cohorts of DCIS single cells. Tumor cells binned into classes after assessing copy number in 500 kb windows with the Gingkgo algorithm.

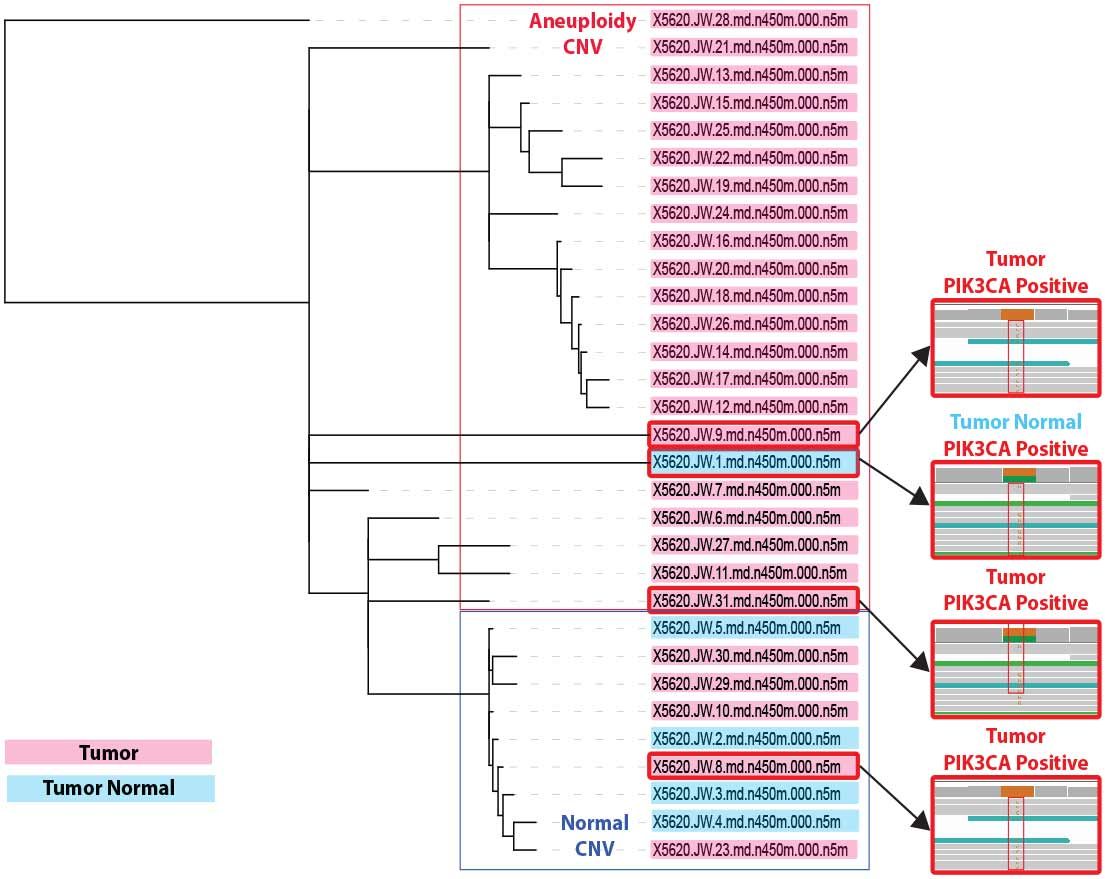
Figure 9. CNV and PIK3CA clonal analysis. A phlyogenetic lineage structure was derived from the DCIS patient cell CNV dataset, onto which we layer PIK3CA H1047R mutation data. DCIS/IDC tumor cells are shown in pink; cells from the ipsilateral normal breast control sampling are presented in blue. Read structures for PIK3CA H1047R are shown (right) and linked to the corresponding CNV clade for that cell.
A second class of single cells contained both Chr. 13 and Chr. 16/17 loss--representing ~20% of the cells (Figure 8C).
A third cohort of cells (~25%) contained these same two CNV alterations plus loss of 11q, another frequently lost region in DCIS (3). These data suggest different clonal populations, defined by CNV, within the tumor milieu (Figure 9) that would not be discernable by bulk sequencing.
Concurrently with CNV analysis, we performed a candidate gene screen for SNVs in genes known to be influential in DCIS (and in breast cancer in general). From this initial screen we identified a H1047R missense mutation in the kinase domain of the lipid kinase PIK3CA; a known activating mutation as well as a known hotspot mutation based on The Cancer Genome Atlas data (5). This change was identified in 4 single cells, 3 from the DCIS/IDC singulated tumor sample and in 1 cell derived from the ipsilateral normal breast control.
Intriguingly, we did not detect PIK3CA H1047R in the single cells with pronounced copy number. This suggests distinct mechanisms of oncogenesis. Some cells within the tumor proliferate uncontrollably due to loss of key tumor suppressor regulation, while in other single cells a missense mutation in a key signal transduction node affecting downstream MAPKmediated cell proliferation and AKT-mediated survival signaling is sufficient to drive unchecked growth.
The presence of the PIK3CA H1047R mutation in one cell derived from the ipsilateral normal breast control surgical resection raises the possibility that the tumor/normal boundary may have been breached during specimen collection. Alternatively, we may have identified a rare pre-malignant cell present in normal tissue. The results, taken together lead us to the belief that WGS with PTA will ultimately become diagnostic to determine the clonal architecture that will provide actionable data to clinicians.

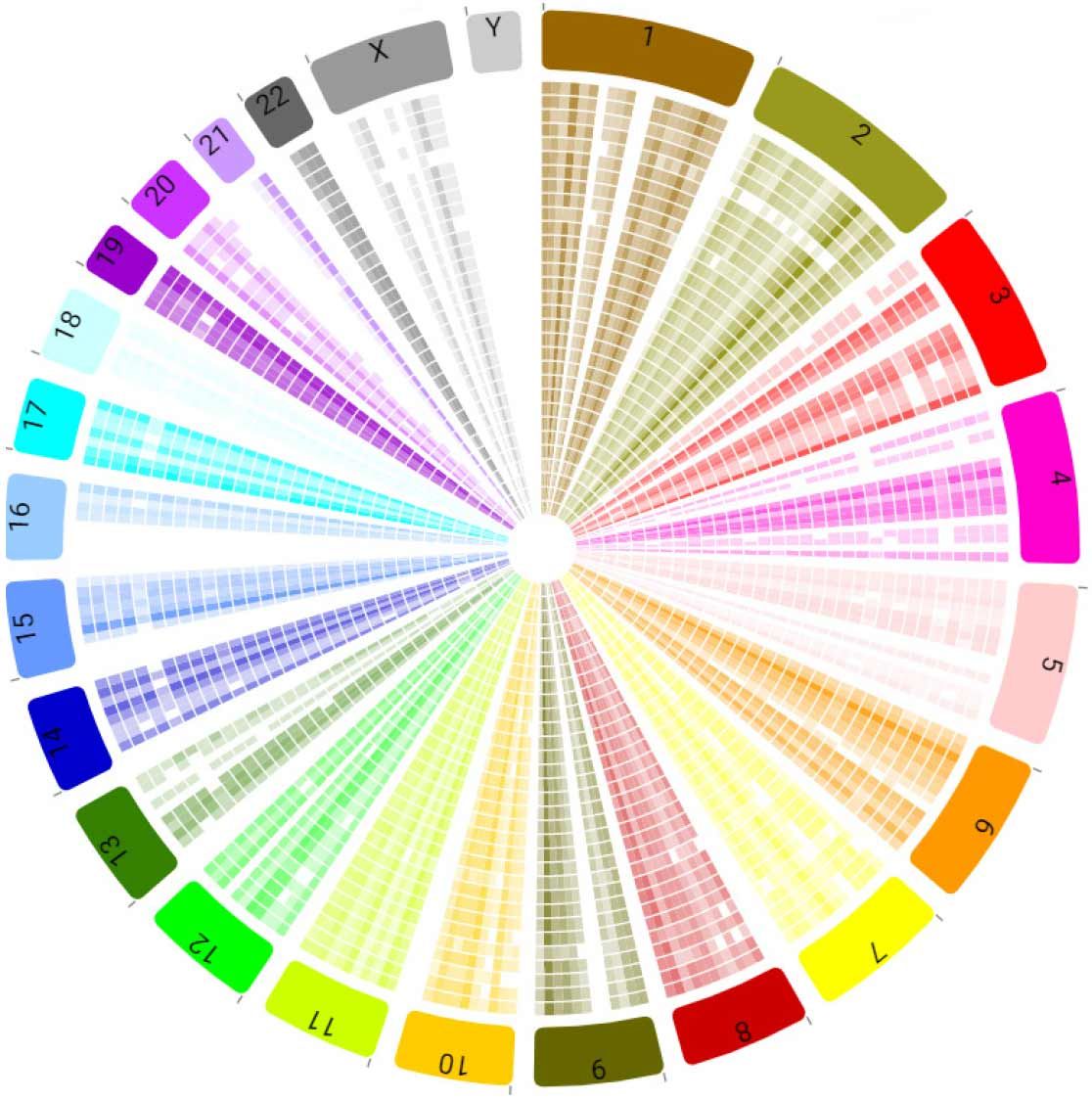
Figure 10. Genomic variation visualized: BaseJumper genomic analysis suite. BioSkryb's BaseJumper™ software facilitates analysis and visualization of genome-wide PTA variant data. Shown is a Circos plot presenting variant density of the 31 patient cells (each concentric ring) employed in this study, and parsed by chromosome number (colors). An outlier under-performing cell is readily visualized by consistent white gaps in variant density, while regional differences in the variant density window of 12.5 Mb can be ascertained.
Summary
This study underscores the need to assess intratumoral clonal heterogeneity, at the single cell level, to both discover new molecular events driving pathology, and potentially drug discovery, as well as identify currently actionable variants. In just 24 single cells we were able to uncover at least 8 different genotypes of cells within the tumor (inclusive of CNV and SNV). Of chief interest here is the exploration of novel variants exclusive of common and characterized CNV/SNV changes that may be contributing to the DCIS transition in this patient and others. The power of uniform whole genome sequencing of single cells that is delivered by PTA allows researchers and clinicians to identify SNV outside of the exonic space, allowing for the contributions of elements like promoters, enhancers, insulators and splice site regulators to be elucidated and understood. Ultimately, studies like this one lay the groundwork for future drug development by identifying candidate targets. We are currently ascertaining non-coding sequence variants with this patient's single cell data to uncover novel variants that might be associated with the DCIS to IDC transition utilizing BaseJumper, BioSkryb Genomics' cloud-based genomic analysis/visualization software (Figure 10).
References:
- Cowell CF, et al. Mol Oncol. 2013 doi: 10.1016/j. molonc.2013.07.005.
- Gonzalez V, et al. PNAS June 15, 2021 118 (24) e2024176118; https://doi.org/10.1073/pnas.2024176118
- Gorringe KL, et al. Mod Pathol. 2015 Sep;28(9):1174-84. doi:10.1038/modpathol.2015.75.
- Daley T & Smith AD. Bioinformatics. 2014 doi:10.1093/bioinformatics/btu540
- Jia M, et al. Breast Cancer. 2021 doi: 10.1007/s12282-020-01199-5.


For more information or technical assistance: info@bioskryb.com
TAS-005, 01/2022